At Polaris Quantum Biotech, we’re leveraging quantum computing to accelerate drug discovery. Once a druggable protein target is identified, it becomes a race to discover a preclinical drug candidate for the project. One of the slowest steps in drug discovery is generating a diverse library of starting candidates. In the past, these libraries have been limited to generic options that are not specific to the protein pocket in question, searches of published literature, and unpatentable small molecules. To accommodate for these limitations, companies resort to expensive, iterative wet lab tests that take years to complete. These options generally result in a starting library with a small chemical space, low diversity, and high bias.
To solve this problem, Polaris Quantum Biotech treats drug design as a multi-objective optimization problem that a quantum annealing computer can solve. Because quantum annealing technology can search unprecedented chemical spaces at high speed, it is possible to design a combinatorial solution space of 1030 and then search it for the combinations of fragments that satisfy a given set of criteria. For example, the output libraries of selected molecules will have structures that are most complementary to the specific binding pocket in question, maximize binding, minimize toxicity, increase metabolic stability, and ensure that all are readily synthesizable.
We developed Quantum-Aided Drug Design (QuADD), which can be used in collaboration with PolarisQB scientists or as a Software as a Service (SaaS) platform for library generation. With the input of a 3D structure of the protein binding pocket and bound ligand, QuADD generates a multi-billion search space to find lead-like hits that are novel, bioavailable, and synthesizable. This output, which includes candidates for best-in-class, first-in-class, and scaffold-jumping structures, provides an enriched starting library of 1K-10K molecules (depending on binding-pocket size) for your drug discovery project. This number of molecules is great enough in size to include a range of options, but small enough that it can be handled with traditional computer-aided drug design programs or high-throughput screening. QuADD includes key parts of our established pipeline that have been discussed in Nature Biopharma Dealmakers [1], Ars Technica [2], and Quantum Insider [3].
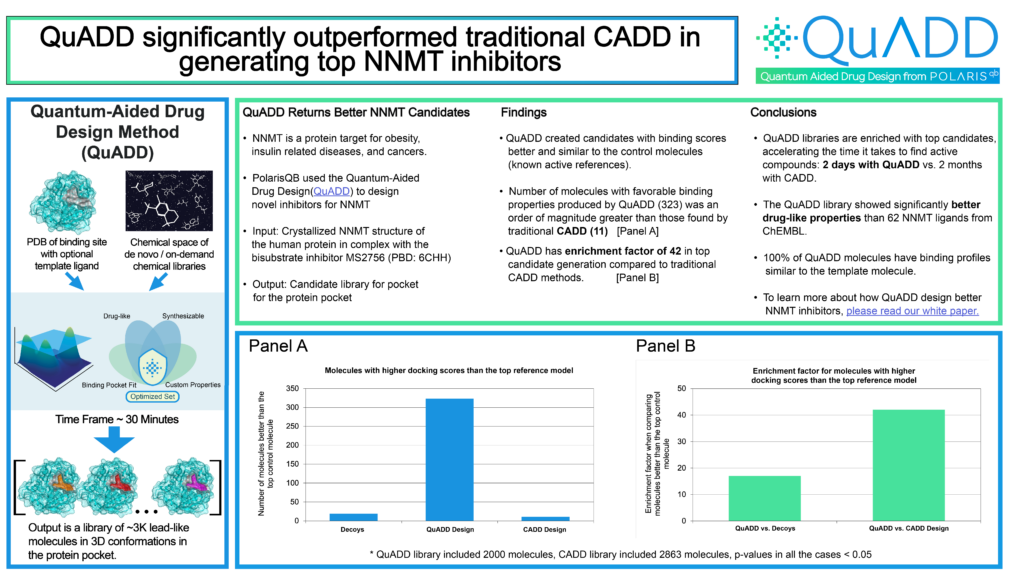
This paper details a case study for the target Nicotinamide N-Methyltransferase (NNMT), which is an important and emergent target for obesity, insulin-related diseases, and different types of cancer [4]. As the starting point for our QuADD calculations, we selected the human crystallized NNMT protein structure in complex with the bisubstrate inhibitor MS2756 from the Protein Data Bank (PDB code: 6CHH) [5]. Results demonstrate that QuADD recovers clinically-proven fragments, discriminates between control and decoy molecules, and identifies molecules that are energetically favorable in the pocket, have a rational binding profile, and present drug-like properties. Taken together, QuADD can facilitate drug discovery by providing enriched small molecule libraries for specific protein pockets, an advantage that lowers the risk and shortens the time to clinical testing