Introduction
- Computational drug design typically requires a well-defined protein-ligand complex. Frequently, only apo structures, which do not have bound ligands, are available. This limits exploration of druggable targets to sites that are open and relatively rigid in their apo states.
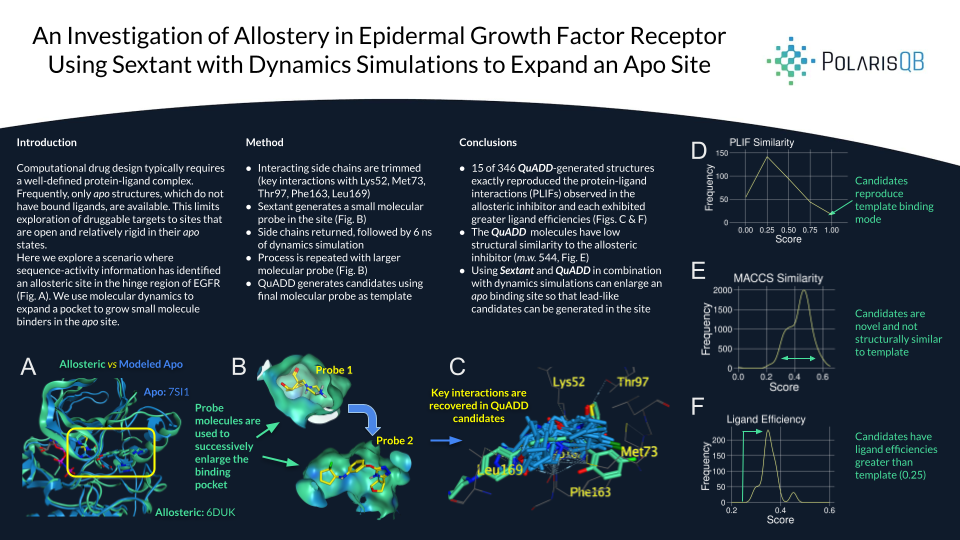
- Here we explore a scenario where sequence-activity information has identified an allosteric site in the hinge region of EGFR (Fig. A). We use molecular dynamics to expand a pocket to grow small molecule binders in the apo site.

Method
- Interacting side chains are trimmed (key interactions with Lys52, Met73, Thr97, Phe163, Leu169)
- Sextant generates a small molecular probe in the site (Fig. B)
- Side chains returned, followed by 6 ns of dynamics simulation
- Process is repeated with larger molecular probe (Fig. B)
- QuADD generates candidates using final molecular probe as template

Conclusions
- 15 of 346 QuADD-generated structures exactly reproduced the protein-ligand interactions (PLIFs) observed in the allosteric inhibitor and each exhibited greater ligand efficiencies (Figs. C & F)
- The QuADD molecules have low structural similarity to the allosteric inhibitor (m.w. 544, Fig. E)
- Using Sextant and QuADD in combination with dynamics simulations can enlarge an apo binding site so that lead-like candidates can be generated in the site