
Quantum AI is an emerging approach that uses quantum computers to run or enhance AI and machine-learning algorithms, aiming to exploit qubits, superposition, and entanglement to process information in ways classical hardware cannot. It is an exciting avenue of research because, if it works at scale, it could dramatically speed up training of large models, handle far more complex data and optimization problems, and unlock new capabilities in areas like drug discovery, finance, logistics, and language understanding that are currently bottlenecked by classical computing limits.Annealing quantum computers excel at combinatorial optimization, making them well-suited for supercharging molecular design, screening, and optimization workflows, including tasks such as selecting the optimal fragments used to build molecular candidates, designing core structures for these molecules, or deciding which molecules to substitute in or out from enormous chemical libraries. These capabilities transform drug discovery into a tailorable design problem.
At PolarisQB, we’re constantly exploring how quantum computing can solve real challenges in drug discovery. In our latest research, we applied quantum machine learning techniques to predict whether drug compounds are metabolized by CYP3A4, the liver enzyme responsible for breaking down roughly half of all marketed drugs. Accurately predicting CYP3A4 substrate status is critical for assessing drug-drug interactions and ultimately determining whether a candidate molecule can safely advance through clinical development. This research was conducted as part of the TD Commons ADMET benchmark challenge on CYP3A4 substrate prediction (https://tdcommons.ai/benchmark/admet_group/15cyp3a4s/).
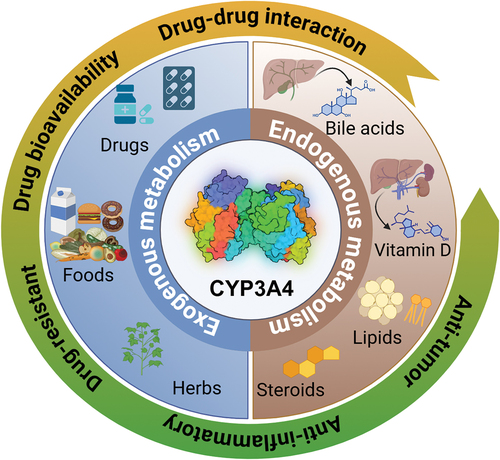
Image taken from:
https://www.tandfonline.com/doi/full/10.1080/13543776.2025.2470294
Our approach, inspired by recent work by Simen et al. (“Data-informed Qubit Assignment with a Variable Arity Quantum Convolution,” arXiv:2510.13807), uses mutual information (MI) analysis to design quantum circuits that capture meaningful relationships among molecular features. Following the work of Simen et al., we identify which pairs and triplets of molecular descriptors share strong statistical dependencies, then encode those relationships into the quantum circuit’s entanglement structure. This data-driven methodology ensures that quantum encoding focuses computational resources on feature interactions that are actually relevant to the prediction task.
We evaluated 48 hyperparameter configurations across a dataset of 668 drug molecules described by 88 molecular descriptors, using 5-fold stratified cross-validation with CatBoost as the classifier. Simpler quantum circuits consistently outperformed more complex ones. The optimal configuration used direct MI coupling, a short evolution time (T=0.5), and a single Trotter step, achieving an ROC-AUC of 0.675 with a standard deviation of just 0.019 across folds, compared to 0.666 ± 0.061 for the classical baseline. This represents a 70% reduction in prediction variance alongside a modest gain in accuracy. The finding aligns with emerging best practices in quantum machine learning: let the data guide the circuit design, and avoid unnecessary complexity.
Our results demonstrate another key advantage of quantum-augmented features: stability. While our quantum-enhanced model achieved a modest 1.4% improvement in ROC-AUC over the classical baseline, the more significant finding was a 70% reduction in prediction variance across cross-validation folds. In practical terms, this means that the model produces consistent predictions regardless of the subset of the training data on which it is presented. This is a crucial property for any system intended for production deployment in drug discovery pipelines.
This work builds on recent research in data-informed quantum circuit design while introducing practical improvements, such as cross-fold stability filtering, to ensure robust feature selection. As quantum hardware continues to mature, techniques like MI-guided Hamiltonian encoding offer a principled path toward quantum advantage in molecular property prediction: not through raw performance gains alone, but through the kind of reliable, consistent predictions that drug discovery demands.